Udover at et lægemiddel skal designes til at binde til target, så er det vigtigt tage højde for, at lægemidlet skal kunne klare en masse udfordringer på dets vej gennem kroppen. Lægemidlet skal modstå mavesyren og fordøjelsesenzymerne, blive optaget over tarmen, modstå metaboliske enzymer i leveren samt undgå at blive ophobet i fedtvæv. Det skal have en tilpas levetid, som ikke er for lang og heller ikke er for kort. Alt dette kaldes i den farmaceutiske industri for ADME, som står for Absorption, Distrubution, Metabolisering og Ekskretion.
Anatomi
Når man designer et lægemiddel må man først og fremmest vide noget om, hvilke udfordringer et lægemiddel møder på sin vej gennem kroppen. Når et lægemiddel indtages oralt, kommer det igennem hele fordøjelseskanalen. Fordøjelseskanalen kan inddeles i mund, spiserør, mavesæk og tarm. I munden bliver lægemidlet udsat for de forskellige enzymer, der findes i vores spyt. Der er dog tale om ret små mængder af enzymer sammenlignet med den mængde af enzymer, som lægemidlet bliver udsat for senere.
Herefter kommer lægemidlet videre til maven. I maven vil lægemidlet blive udsat for den meget sure mavesyre (pH = 1 – 3), der meget hurtigt kan nedbryde lægemidlet. Overlever lægemidlet dette, sendes det videre til tarmene, hvor det bliver udsat for en stor mængde fordøjelsesenzymer. Disse fordøjelsesenzymer nedbryder normalt den mad, vi indtager. Hvis lægemidlet har overlevet disse ”angreb”, er det nu tid til at krydse epitelcellelaget i tarmen for at trænge ind til blodbanen. For at kunne blive optaget i blodbanen, skal lægemidlet have den rette polaritet. Dette inkluderer, at lægemidlet skal være i stand til at kunne krydse cellemembraner, som er hydrofobe, men det skal også kunne befinde sig i blodet, som er hydrofilt.
Efter at lægemidlet er optaget i blodet, vil det blive transporteret til leveren. I leveren findes en række enzymer, som har til opgave at ændre den kemiske struktur af stoffer, som er fremmede for kroppen. Dette kaldes for metabolisering af stofferne. Når stofferne er ændret, bliver de lettere udskilt med urinen. At lægemidlet passerer leveren, før det når ud til vævet, kaldes for first pass effekt. Hvis lægemidlet nemt bliver metaboliseret, og metaboliseringen sker hurtigt, vil der ikke blive fordelt noget af lægemidlet ud til vævet. Lægemidlet skal derfor have en langsommere metabolisering, så det kan nå at være aktivt.
Absorption
Når et lægemiddel er indtaget oralt og har været gennem hele fordøjelseskanalen, skal det optages henover tarmen. Dette kaldes for absorption af lægemidlet og der kan i absorptionsfasen opstå en række forskellige problemer.
Syre/base kemi
Hvis et lægemiddel er ioniseret kan det ikke optages i kroppen gennem tarmen, fordi ioniserede molekyler ikke kandiffundere passivt over en cellemembran. Det er derfor vigtigt at vide, hvornår et molekyle er ioniseret og hvornår det ikke er, og det er her at syre/base kemi bliver vigtigt.
Der findes flere forskellige definitioner af syrer og baser; fx Brønsted-Lowry– og Lewis-definitionen. Brønsted-Lowry definitionen er den, der bliver brugt mest og er den generelle opfattelse af, hvad syrer og baser er. En Brønsted-Lowry syre er et molekyle, der kan afgive en hydron (ioniseret hydrogen, H+), og en Brønsted-Lowry base er et molekyle, der kan optage en hydron. Et molekyle kan også være begge dele på en gang. Vand er eksempelvis både en syre og en base. Vand kan nemlig afgive en hydron, hvorved det fungerer som en syre:

Reaktion 1. Vands reaktion som en syre.
Vand kan også optage en hydron, og fungerer derved som en base:

Reaktion 2. Vands reaktion som en base.
Når en kemisk reaktion forløber, vil der indstille sig en ligevægt. Dette gælder også for vands reaktion med sig selv. Når en ligevægt har indfundet sig, vil det sige, at reaktionen har nået et bestemt punkt, hvor der er lige meget omdannelse fra reaktant til produkt, som der er omdannelse fra produkt til reaktant. Reaktionshastigheden er derfor lige hurtig frem og tilbage, men der er ikke nødvendigvis lige meget produkt og reaktant.
Som beskrevet ovenfor består vand ikke kun af H2O-molekyler, men også af H+ og OH–. En ligevægt for H2O kan derfor skrives op på følgende måde:

Reaktion 3. Ligevægt for H2O
Ud fra denne ligevægt kan pH beregnes. pH er et udtryk for hvor mange hydroner, der er i blandingen, og dermed hvor sur blandingen er. Helt simpelt tager man den negative logaritme til koncentrationen af hydronerne:

Ligning 1. Formel for udregning af pH
Hvis vand blandes sammen med eksempelvis en syre, vil der også her indstille sig en ligevægt i blandingen. Denne ligevægt afhænger af, hvor stærk eller svag syren er. Hvis det er en stærk syre, vil ligevægten blive forskudt helt mod højre i reaktionsskemaet vist i figur 38 og al den oprindelige syre er omdannet til den konjugerende base.

Figur 38. Viser ligevægten af reaktionen mellem en syre og H2O, hvor H2O reagerer som en base. Formlen for den dertil hørende syrestyrke konstant er desuden angivet.
Styrken af en syre kan bestemmes via syrestyrke-konstanten, Ks. Denne kan beregnes via formlen, der ses i figur 38. Man tager koncentrationen af de produkter, der er på højre side af ligevægtspilene, og dividerer dem med koncentrationen af de reaktanter, der står på venstre side af ligevægtspilene. Vand tages ikke med, da det anses som et opløsningsmiddel, og derfor hverken er en reaktant eller et produkt. Styrken af syren bliver dog for det meste angivet som pKs, hvilket svarer til at tage den negative logaritme til Ks:

Ligning 2. Formel til beregning af pKS.
Når syrestyrken beregnes ud fra ovenstående formel, vil stærke syrer have en lav pKs-værdi, og svage syrer vil have en høj pKs-værdi.
Den samme konstant findes også for baser. Basestyrken er dog sjældent opgivet, og det er oftest syrestyrken for den korresponderende syre til basen, som man kan slå op. Man kan dog heldigvis let regne sig frem til basestyrken ud fra syrestyrke-konstanten via følgende formel:

Ligning 3. Formel til beregning af pKb
Som nævnt tidligere er syre/base kemi vigtig for optagelsen af lægemidler i tarmen. Når svage baser opløses i en blanding der har en pH-værdi der er højere end pK>-værdien, er basen uioniseret. Den svage base kan derfor krydse over tarmens celler, idet kun uioniserede molekyler kan diffundere over celler. I de områder i tarmen hvor stoffer bliver optaget i kroppen, er der en pH-værdi på mellem 6 og 9. Et lægemiddel der har en pKS-værdi på omtrent 8, er en svag base. Så hvis lægemidlet har en pKS-værdi på 8, vil lægemidlet højst sandsynligt kunne optages over tarmen. Syrer skal ligesom baser også være uioniserede for at kunne optages i tarmen. Dette er muligt for svage syrer når pH i tarmen er under pKS for lægemidlet. Det gælder derfor om at have et lægemiddel med en pKS-værdi på mellem 6 og 8. Der findes i dag en del lægemidler på markedet, der indeholder aminer. Dette er der to gode grunde til. For det første binder aminer godt til mange targets, men vigtigst er, at aminerne er gode til at blive optaget i kroppen, idet de har en pKS-værdi, der svinger mellem 6 og 8.
Partitionskoefficienten P
Nogle lægemidler er så hydrofobe at de vil blive fanget i cellemembraner og ophobe sig i fedtvæv. Andre lægemidler er så hydrofile at de ikke vil være i stand til at krydse cellemembraner og derfor ikke vil kunne blive optaget oralt. En måde at vurdere, hvor hydrofobt eller hydrofilt et lægemiddel er, dvs. hvor opløseligt lægemidlet er i enten fedt eller vand er ud fra partitionskoefficienten P. Partitionskoefficienten bestemmes ved at komme sit molekyle ned i en blanding af 50 % octanol og 50% vand. I et sådant system vil hydrofobe molekyler befinde sig i octanollaget, mens de molekyler, der ikke er hydrofobe, men derimod er hydrofile, vil befinde sig i vandlaget:
![P=\frac{[Molekyle]_{octanol}}{[Molekyle]_{vand}}](https://www.biotechacademy.dk/wp-content/ql-cache/quicklatex.com-4265bccc4b048a6b00a6f33cb61b5255_l3.png "Rendered by QuickLaTeX.com")
Ligning 4. Formel til beregning af partitionskoefficienten P. Firkantede parenteser bruges til at angive koncentrationer i de to faser, octanol og vand.
Som ved de fleste andre fordelingskoefficienter, angiver man ofte P som logP værdien. Hydrofobe molekyler vil have en høj logP-værdi, og hydrofile molekyler vil have en lav logP-værdi.
Man kan også få et computerprogram til at beregne en estimeret værdi, kaldet ClogP (calculated logP). Ligesom enkelte funktionelle grupper, såsom aminer, kan være på en ioniseret og uioniseret form, kan hele lægemidlet inklusiv dets funktionelle grupper også være delvist ioniseret eller uioniseret. LogP-værdien angiver kun hydrofobiciteten af de uioniserede molekyler. Hvis man ønsker at måle på de ioniserede molekyler, skal man i stedet anvende logD. I dette projekt er det dog kun logP vi skal bruge.
Lipinskis regel af 5
For at kunne vurdere om et lægemiddel er muligt at optage oralt, og kunne absorberes over tarmepitelcellelaget, og dermed give lægemidlet en god biotilgængelighed, er en vigtig tommelfinger-regel blevet udviklet. Denne tommelfinger-regel kaldes for Lipinski’s regel af 5, fordi alle punkterne går op i 5.
Lægemidlet skal:
- Have en molekylvægt under 500 Da
- Ikke have flere end 5 hydrogenbindingsdonorer (HBD)
- Ikke have flere end 10 hydrogenbindingsacceptorer (HBA)
- Have en logP under +5
Det skal nævnes, at selvom der er to ledige elektronpar på det samme atom, som eksempelvis på et dobbeltbundet oxygenatom, så tælles disse ledige elektronpar kun som én HBA ifølge Lipinski’s regel.
Hvis et molekyle har logP > 5, vil molekylet være for hydrofobt til at kunne opløses i vand. Molekylet er derfor heller ikke opløseligt i blodet, hvilket medfører at molekylet bliver tilbageholdt i cellemembranerne. Hvis logP til gengæld er meget lav for et molekyle <1, vil molekylet være for hydrofilt til at kunne krydse cellemembraner. Molekylet vil derfor ikke blive optaget i kroppen, og det bliver udskilt af kroppen uden at have udført sin virkning.
Lægemidler som har mange grupper, der virker som HBA eller HBD, er gode til at lave hydrogenbindinger med vand. For at et molekyle kan krydse en cellemembran, skal det ikke være bundet til vand og derfor skal der brydes en masse hydrogenbindinger mellem vand og molekylet. At bryde disse hydrogenbindinger mellem molekylet og vand kræver energi, og derfor skal antallet af HBA og HBD i lægemidlet være lavt for at mindst mulig energi skal bruges i transporten af molekylet over cellemembranen.
Det er ikke et krav, at et godt lægemiddel opfylder alle Lipinski’s regler. Reglerne er blevet udviklet ved at sammenligne de lægemidler, der findes på markedet for at finde nogle ligheder mellem dem. Der findes derfor også lægemidler, der falder udenfor Lipinski’s regler. Eksempelvis har ciclosporin, der er et lægemiddel, som sænker immunforsvarets aktivitet, en molekylvægt på 1203 Da, hvilket ligger langt over de 500 Da, som Lipinski’s regel angiver. Molekylet har desuden ekstremt mange hydrogenbindingsacceptorer (HBA).
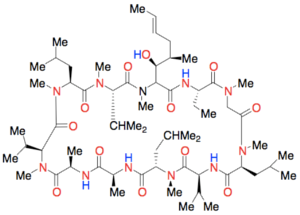
Figur 39. Viser lægemidlet ciclosporin, hvor HBA er vist med rød og HBD med blå.
At et lægemiddel har en høj molekylvægt, er ikke ensbetydende med, at det har en lav oral biotilgængelighed. Jo større et molekyle er, desto flere funktionelle grupper vil der dog også være, som muligvis kan lave hydrogenbindinger, hvilket netop er tilfældet med ciclosporin. Derfor vil en høj molekylvægt oftest medføre lav oral biotilgængelighed.
Distribution
Efter at lægemidlet er blevet optaget i kroppen, skal det fordeles rundt i kroppen og specielt skal det transporteres ud til det område, hvor lægemidlet skal have sin virkning. Først og fremmest bliver lægemidlet fordelt rundt i kroppens blodbaner (efter først at have været en tur i leveren, se under metabolisme afsnit). Fra blodet vil lægemidlet blive optaget af forskellige celler i kroppen. Det kan have forskellige konsekvenser for distributionen af et lægemiddel, alt efter hvilken kemisk struktur molekylet har. Hvis lægemidlet eksempelvis binder godt til de røde blodlegemer i blodet, vil det ikke blive optaget i kroppens væv, hvilket var hensigten.
For at være sikker på, at et lægemiddel bliver ordentligt distribueret, gælder det om at afbalancere hvor hydrofobt lægemidlet er i forhold til, hvor hydrofilt det er. En for høj hydrofobicitet kan have uønskede bivirkninger, da lægemidlet kan ophobe sig i fedtvæv. Fx skal overvægtige patienter, der skal opereres, have en større mængde af nogle bedøvelsesmidler end normalvægtige personer, fordi bedøvelsesmidlet ophober sig i de overvægtige patienters fedtvæv. Når operationen er ovre, og patienten vågner, vil der stadig være en del bedøvelsesmiddel tilbage i fedtvævet. Bedøvelsesmidlet bliver frigivet fra fedtvævet, og kan resultere i, at patienten igen bliver bevidstløs, hvilket er en meget uheldig bivirkning.
Metabolisme
Efter oral indtagelse af et lægemiddel og herefter optagelse i blodbanen, er leveren det første sted, hvortil et lægemiddel bliver ledt hen (first pass effekt). Dette skyldes, at der fra tarmene er en blodåre, der fører direkte til leveren. Denne blodåre kaldes for den portale vene. Et lægemiddel, der indtages oralt, bliver derfor ført til den portale cirkulation, som er blodtransport fra tarmene og direkte til leveren. Hvis man i stedet giver lægemidlet ved i.v. indsprøjtning, vil det komme ind i den systemiske cirkulation. I dette tilfælde vil lægemidlet cirkulere en gang igennem blodbanerne i hele kroppen inden det når til leveren. Der vil ved i.v. indsprøjtning være en stor procentdel af den givne dosis af et lægemiddel, der vil nå sit target, inden det bliver ført til leveren og nedbrudt. Ved indtagelse af lægemidlet oralt er der til gengæld en stor procentdel af lægemidlet, der ikke vil nå sit target førend der er blevet nedbrudt i leveren.
Som nævnt findes der i leveren en række enzymer. Disse enzymer har til opgave at ændre strukturen af lægemidlet, så lægemidlet bliver udskilt fra kroppen. Lægemidler, der er meget polære, vil med det samme blive udskilt fra blodet gennem nyrerne og føres ud i urinen. Upolære molekyler er derimod vanskeligere for kroppen at udskille gennem nyrerne, men via metabolske processer, kan molekylet gøres mere polært, så det kan blive udskilt. Et metaboliseret molekyle kaldes for en metabolit. Når et lægemiddel bliver metaboliseret, vil det ofte miste sin oprindelige effekt. I nogle tilfælde kan metabolitten dog stadig have en smule aktivitet tilbage. Desuden kan ændringen i molekylet føre til dannelsen af toksiske biprodukter, som kan forårsage uønskede bivirkninger. Det er derfor vigtigt at have kendskab til, hvilke metabolitter der kan dannes for at mindske toksiske bivirkninger.
Ethvert nyt lægemiddel skal have gennemgået en metabolittest in vivo, så det kan bestemmes, hvilke metabolitter der dannes, når lægemidlet optages i et komplekst system. Det er dog ikke sikkert, at de metabolitter der dannes i forsøgsdyr er de samme, som vil blive dannet i mennesker. Man kan derfor aldrig være 100 % sikker på, hvilke metabolitter der dannes i mennesker, før en metabolittest i mennesker er udført.
I artiklen ”Organisk kemi og lægemidler” er det blevet beskrevet, hvordan typen af isomer af et lægemiddel er vigtig i forhold til, hvordan molekylet påvirker sit target. Det er dog ikke kun i forhold til påvirkning af target, at typen af isomeren er vigtig. Også i nedbrydningen eller ændringen af lægemidlet er stereokemien vigtig. De enzymer, der metaboliserer lægemidlet, genkender også kun én bestemt isomer. Hvis et lægemiddel består af to forskellige isomerer af det samme stof, vil der muligvis også blive dannet to forskellige metabolitter katalyseret af to forskellige enzymer. Den ene isomer kan være relativ harmløs, mens den anden kan være særdeles toksisk. Derfor skal begge isomerer testes in vitro hver for sig for at bestemme, hvilke metabolitter der dannes ud fra dem. Der vil være mere arbejde i, at bestemme hvilke metabolitter der dannes in vitro, og hvilke toksiske bivirkninger der kan være fra den anden isomer og det er derfor bedst kun at have en enkelt isomer i sit lægemiddel. Det er derfor er vigtigt at designe en fremgangsmåde til fremstillingen af lægemidlet, hvor der kun dannes den ene isomer (stereospecifik syntese).
Der findes to forskellige former for metabolisme, fase I og fase II metabolisme, som kan forløbe i leveren. Fase I metabolisme er oxidationsreaktioner, der bliver katalyseret af cytochrom P450 enzymer (CYP). CYP enzymer har til opgave at gøre det pågældende molekyle mere polært. Fase II metabolisme er konjugationsreaktioner, hvor der til en polær gruppe på molekylet bliver bundet et ekstra molekyle, således at hele molekylet bliver mere polært, end det i forvejen var. Begge typer metabolisme vil medvirke til, at stoffet bliver endnu hurtigere udskilt fra kroppen.
Fase I
CYP enzymer er hæmproteiner, hvilket vil sige, at de indeholder en hæmgruppe samt jern. De tilhører gruppen af monooxygenaser og splitter O2 der er tilstede i leveren, således at det ene oxygenatom overføres til lægemidlet, som derved bliver oxideret, mens det andet oxygenatom bindes til to hydrogenatomer og danner vand. For at lægemidlet kan blive oxideret, er der stoffer der nødvendigvis må blive reduceret. Derfor kræver CYP enzymerne tilstedeværelse af coenzymet NADPH. Når lægemidlet bliver oxideret, bliver NADPH reduceret til NADP.

Figur 40. Viser et lægemiddel der bliver oxideret af cytochrom P450, samtidig med at NADPH bliver reduceret til NADP.
Der findes mindst 33 forskellige CYP enzymer, som kan inddeles i forskellige undergrupper, der hver især varetager en bestemt reaktion. Det første trin i de fleste fase I metaboliseringsreaktioner er tilførslen af en alkohol gruppe. Denne kan indføres på forskellige positioner i et lægemiddel-molekyle, som illustreret i figur 43. Alt efter hvor alkoholgruppen bliver tilført, bliver OH-gruppen ofte oxideret. Denne oxidation kan enten være til en keton eller en aldehyd. Hvis OH-gruppen er blevet omdannet til en aldehyd, vil aldehyden blive oxideret videre til en carboxylsyre. En af de funktionelle grupper, man skal være særligt opmærksom på, er methyl grupper (CH3), da disse meget nemt oxideres til carboxylsyrer via sekundære alkoholer. Det kan ses på figur 6, at nitrogenholdige funktionelle grupper også bliver oxideret.
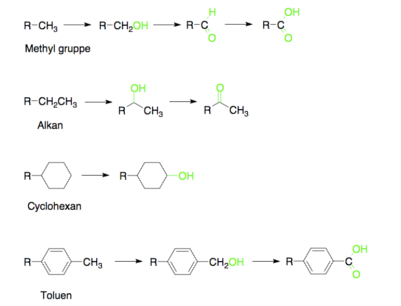
Figur 41. Viser et udvalg af forskellige funktionelle grupper der bliver oxideret af CYP enzymer i fase I metabolisme og hvad de forskellige grupper oxideres til.
Fase II
De fleste enzymer, der står for fase II metabolisme, tilhører enzym-gruppen transferaser. Transferaser er enzymer, der overfører en funktionel gruppe fra ét molekyle til et andet. Der konjugeres en funktionel gruppe til lægemidlet, og fase II metabolisme kaldes derfor for konjugationsreaktioner. De funktionelle grupper, der overføres, kan være mange forskellige. Et eksempel på en funktionel gruppe der kan overføres i en fase II metabolisering, er dannelsen af O-glucuronider, ud fra funktionelle grupper indeholdende OH-grupper. Ofte sker en fase II metabolisering efter en fase I metabolisering, således at der i fase I dannes en OH-gruppe, hvorpå der i fase II påsættes en funktionel gruppe.
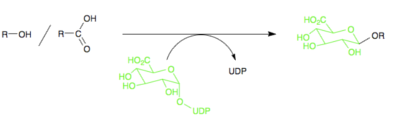
Figur 42. Viser dannelsen af en O-glucoronid ud fra et lægemiddel indeholdende en OH-gruppe, som er blevet påsat i en fase I metabolisering.
Det kan ud fra figur 44 ses, at der ved dannelsen af et O-glucoronid bindes en stor gruppe til lægemidlet, og bindingen af denne gruppe gør lægemidlet meget mere polært.
Hvis lægemidlet indeholder en carboxylsyre, eller hvis en carboxylsyre er blevet dannet under fase I metabolisering, vil der kunne konjugeres en aminosyre fast til lægemidlet. I mennesker er det ofte glutamin, der påsættes.
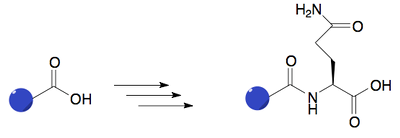

Figur 43. Viser hvordan aminosyrer (i dette eksempel glutamin) bliver påsat via tre trin i en fase II metabolisering.
Der findes også en række andre former for metabolisme af lægemidlet, som ikke foregår i leveren. Der er bl.a. en række oxidative enzymer (ligesom CYP enzymerne) fordelt rundt i kroppens væv, som også indgår i fase I metabolisme af forskellige lægemidler. Desuden foregår der i blodet ikke-enzymatisk, men derimod kemisk nedbrydning af forskellige funktionelle grupper. En af de mest fremtrædende kemiske nedbrydninger er omdannelsen af estre til carboxylsyrer og alkoholer. Denne funktion er meget benyttet ift. nedbrydningen af prodrugs til det aktive lægemiddel, som er beskrevet i artiklen ”Optimering af lægemidlet”. Det kan derfor være svært, at opnå en lang levetid i kroppen for et lægemiddel, der indeholder en ester-gruppe, da det hurtigt nedbrydes i blodet.
Ekskretion
De fleste lægemidler vil blive udskilt via nyrerne igennem urinen. Op til 15% af mængden af et lægemiddel kan dog udskilles via sved. Nogle lægemidler kan desuden udskilles via lungerne, hvis stoffet er en gasart.
Forklaringen på, hvorfor polære stoffer bliver udskilt bedre end upolære stoffer i urinen, ligger i måden, hvorpå nyrerne virker:
Det blod, der kommer fra leveren, bliver samlet i nyrerne. Her bliver det filtreret, så blodlegemer og blodplader ikke bliver udskilt, mens alle ”affaldsstofferne” bliver skilt fra. Det er dog kun en filtrering, så både polære og upolære stoffer kan passere ind i nyrerne, og derved blive tilbageholdt i kroppen. Herefter bliver urinen opkoncentreret, fordi der findes nogle små porer, aquaporiner, i nyrerne, der kan genoptage vandet fra urinen tilbage til kroppen. Udover at vand optages af aquaporiner, bliver en del stoffer genoptaget gennem cellerne i nyrerne. Fordi stofferne genoptages gennem cellernes membraner, skal de være ret upolære/hydrofobe for at kunne trænge igennem. De polære stoffer kan derfor ikke komme igennem, og de vil blive udskilt hurtigt. De upolære stoffer vil blive genoptaget, og de kan cirkulere rundt i kroppen endnu en gang.
Optagelsesformer
Et lægemiddel kan indtages på flere forskellige måder. De vigtigste indtagelsesformer er oralt, ved injektion, inhalering eller gennem tarmen og huden. Oral indtagelse i pilleform er langt den mest anvendte indtagelsesform, idet det er den letteste måde for patienten at indtage sin medicin på. Der er større chance for, at patienten gennemfører sit behandlingsforløb sammenlignet med et forløb, hvor patienten eksempelvis flere gange om dagen skal have sprøjtet lægemidlet ind under huden.
Hvis patienten lider af en alvorlig sygdom, og en oral optagelsesform ikke er mulig, er man nødt til at finde en anden måde, hvorpå lægemidlet kan administreres. Nogle patienter har problemer med at sluge piller, enten fordi de ikke er ved bevidsthed eller kaster meget op, og andre optagelsesformer end den orale er derfor at foretrække. Desuden er der mange børn, der har problemer med at sluge piller, og derfor er optagelse gennem tarmen, via en stikpille, meget benyttet til børn.






















")
")


")
")
")
")

























